Общност
Ретинобластомът (Rb) е злокачествен очен тумор, който се развива от клетките на ретината. Този рак може да се появи на всяка възраст, но началото е най -често в ранна детска възраст преди петгодишна възраст.

Ракът в детска възраст е агресивен: ретинобластомът може да се разпространи в лимфните възли, костите или костния мозък. Рядко засяга централната нервна система (мозък и гръбначен мозък).
Около 90% от децата с ретинобластом имат положителна прогноза (вероятност за излекуване), при условие че диагнозата е ранна и лечението е започнало преди разпространението на рака. Когато е възможно, целта на медицинската намеса е да се запази зрението на пациента.
Причини
Поредицата от събития, водещи до появата на тумор, е сложна. Това започва, когато клетките в ретината развият мутация (или делеция), включваща RB1 туморен супресорен ген, разположен в q14 лентата на хромозома 13 (13q14).
Всяка клетка обикновено има два RB1 гена:
- Ако поне едно копие на гена работи правилно, ретинобластом не възниква (но рискът се увеличава);
- Когато и двете копия на гена са мутирани или липсват, настъпва неконтролирана клетъчна пролиферация.
В много случаи не е ясно какво точно предизвиква промени в гена RB1 (спорадичен ретинобластом); те могат да бъдат резултат от случайни генетични грешки, които възникват например по време на размножаването и клетъчното делене. Известно е обаче, че генетичните аномалии, лежащи в основата на ретинобластом, също могат да бъдат предадени от родители на деца, с автозомно доминантно наследяване. Това означава, че ако родител носи мутирал (доминиращ) ген, всяко дете ще има 50% шанс да го наследи и 50% шанс да има нормален генетичен състав (рецесивни гени).
- Случайна клетка инактивира единственото си нормално копие на гена RB1 (едно копие вече е мутирало);
- Загубата на двете копия на RB1 води до „прекомерна пролиферация на ретината.
- Случайна клетка инактивира един от нормалните си RB1 гени;
- Второто копие на гена RB1 е инактивирано;
- Загубата на двете копия на RB1 предизвиква прекомерна клетъчна пролиферация, която води до ретинобластом.
Генетични и молекулярни характеристики
- Ретинобластомът е първият тумор, който е пряко свързан с „генетична аномалия (делеция или мутация на q14 лентата на хромозома 13).
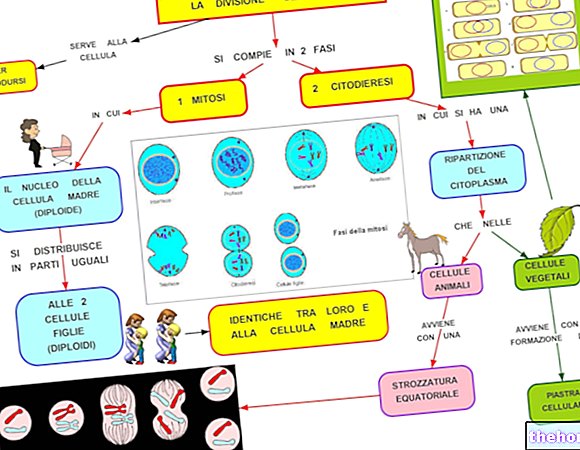
- RB1 кодира протеина pRb, който играе ключова роля в клетъчния цикъл: той позволява репликация на ДНК и прогресия на клетъчния цикъл, тъй като участва в контрола на транскрипцията на гените на S фаза (G1 → † "S).
- В допълнение към ретинобластома, генът RB1 се инактивира при рак на пикочния мехур, гърдата и белия дроб.
Наследствен ретинобластом
Децата с наследствен ретинобластом са склонни да развият болестта в по -ранна възраст от спорадичните случаи. Освен това, тези деца са изложени на повишен риск от други неочни ракови заболявания, тъй като аномалията в гена RB1 е вродена (т.е. присъства от раждането) и засяга всички клетки в тялото (известни като мутация на зародишната линия), включително тези на двете. ретини: Поради тази причина децата с наследствена форма често имат двустранен ретинобластом, а не само едно око.
Симптоми
За да научите повече: Симптоми на ретинобластом
Най-честият и очевиден признак на ретинобластом е анормалният вид на зеницата, който представлява сиво-бяло отражение, когато е ударено от лъч светлина (левкокория или амавротичен котешки рефлекс). Други признаци и симптоми включват: намалено зрение, болка в очите и зачервяване и забавяне на развитието. Някои деца с ретинобластом могат да развият кривогледство (неправилно подредени очи); в други случаи е възможно да се открие неоваскуларна глаукома, която след известно време може да причини уголемяване на окото (буфталмо).
Раковите клетки могат допълнително да нахлуят в окото и други структури:
- Вътреочен ретинобластом. Ретинобластомът може да се определи като вътреочен, когато туморът е изцяло разположен вътре в окото. Неоплазмата може да се открие само в ретината или да засегне и други части, като хороидеята, цилиарното тяло и част от зрителния нерв. Следователно, вътреочният ретинобластом не се разпространява в тъканите около външната страна на окото.
- Екстраокуларен ретинобластом. Туморът може да се размножава, за да засегне тъканите около окото (орбитален ретинобластом). Ракът може да се разпространи и в други части на тялото, като мозъка, гръбначния стълб, костния мозък и лимфните възли (метастатичен ретинобластом).
Наличието на орбитално разширение, увеално засягане и инвазия на зрителния нерв са известни рискови фактори за развитието на метастатичен ретинобластом.
Диагностика
В случай на положителна фамилна анамнеза, пациентът се подлага на редовни очни прегледи за скрининг на рак. Ако вроденият ретинобластом е двустранен, той обикновено се диагностицира през първата година от живота, докато когато засяга само едното око, наличието на тумора може да бъде потвърдено на около 18-30 месечна възраст.
Клиничната диагноза на ретинобластом се установява чрез изследване на фундуса. Технологиите на образна диагностика могат да се използват за потвърждаване на диагнозата, определяне на стадирането на тумора (къде е, колко широко е разпространено, дали засяга функциите на други органи в тялото и т.н.) и да се определи дали лечението е било ефективно . Изследванията могат да включват ултразвук, компютърна томография (КТ) и ядрено -магнитен резонанс (ЯМР).
Молекулярно-генетичната диагноза е възможна чрез идентифициране на мутацията на гена RB1. Цитогенетичният анализ (т.е. на хромозомите) на лимфоцитите в периферната кръв се използва за откриване на делеции или пренареждания, включващи хромозома 13 (13q14.1-q14. 2) .
Лечения
В случай на ретинобластом могат да се използват няколко възможности за лечение.
Целите на лечението са:
- Елиминирайте тумора и спасете живота на пациента;
- Запазете окото, ако е възможно;
- Запазете зрението колкото е възможно повече;
- Избягвайте развитието на други видове рак, които също могат да бъдат причинени от лечението, особено при деца с наследствен ретинобластом.
Прогнозата (вероятността за възстановяване) и възможностите за лечение зависят от следните фактори:
- Етап на тумора;
- Възрастта на пациента и общото здравословно състояние;
- Местоположение, размер и брой туморни огнища;
- Разпространението на рака в други области освен очната ябълка
- Колко вероятно е зрението да се запази в едното или в двете очи.
Повечето случаи на ретинобластом се диагностицират рано и успешно се лекуват, преди ракът да може да метастазира извън очната ябълка, което води до процент на излекуване над 90%.