Общност
Синдромът на Crouzon е рядко генетично заболяване, което определя наличието на краниосиностоза и други доста особени аномалии на лицето.
Появата му е причинена от определени промени в ДНК, която съставлява гените FGFR2 и FGFR3; тези генетични елементи участват в процеса на съзряване на костите по време на ембрионалното развитие.

Терапията се състои от поредица хирургични интервенции, насочени към разрешаване на най -важните и най -опасни симптоми.
В момента прогнозата често е положителна.
Преглед на генетиката
Преди да продължите с описанието на синдрома на Crouzon, е полезно да прегледате някои основни концепции за генетиката.
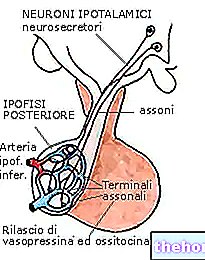
Какво е ДНК? Това е генетичното наследство, в което са записани соматичните черти, предразположенията, физическите качества, характера и т.н. на жив организъм. Той се съдържа във всички клетки на тялото, имащи ядро, като се крие в това.
Какво представляват хромозомите? Според определението хромозомите са структурните единици, в които е организирана ДНК. Човешките клетки съдържат в ядрото си 23 двойки хомоложни хромозоми (22 от автозомния несексуален тип и една двойка от сексуалния тип); всяка двойка е различна от друга, тъй като съдържа специфична генна последователност.
Какво представляват гените? Те са кратки участъци или последователности на ДНК с фундаментално биологично значение: от тях всъщност произтичат протеини или биологични молекули, фундаментални за живота. В гените има „написана“ част от това кои сме и кои ще станем.
Всеки ген присъства в две версии, алелите: един алел е от майчин произход, следователно се предава от майката; другият алел е с бащин произход, поради което се предава от бащата.
Какво е генетична мутация? Това е грешка в последователността на ДНК, която образува ген. Поради тази грешка полученият протеин е или дефектен, или напълно липсва.И в двата случая ефектите могат да бъдат вредни както за живота на клетката, в която се случва мутацията, така и за този на организма като цяло. Вродените заболявания и неоплазмите (т.е. туморите) принадлежат към една или повече генетични мутации.
Синдромът на Крузон също
Синдромът на Крузон е рядко генетично състояние, характеризиращо се с краниосиностоза и „неестествено развитие на определени елементи на лицето, включително очите, носа, челюстта и челюстта“.
Това е вродено заболяване, чиито типични характеристики могат да се проявят още през първите моменти от живота.
ЗНАЧЕНИЕ НА КРАНИОЗИНОСТОЗИ
Краниосиностоза е терминът, чрез който лекарите се позовават на преждевременното сливане на един или повече черепни конци.

От сайта: thecraniofacialcenter.com
Черепните конци са влакнестите стави, които съединяват костите на черепния свод (т.е. челната, темпоралната, париеталната и тилната кост).
При нормални условия сливането на черепните конци се случва в постнаталния период (някои процеси дори завършват на 20-годишна възраст). Този дълъг процес на сливане позволява на мозъка да расте и да се развива адекватно.
Ако, както в случая с краниосиностози, сливането се извършва твърде рано - следователно по време на пренатален, перинатален * или ранен детски живот - мозъчните елементи (мозък, малък мозък и мозъчен ствол) и някои сетивни органи (по -специално очите) претърпяват „промяна на формата и растежа.
* Перинатален термин се отнася за периода от живота, който преминава от 27 -та гестационна седмица до първите 28 дни след раждането.
ПРОИЗХОД НА ИМЕТО
Синдромът на Crouzon дължи името си на френския лекар Octave Crouzon, който има заслугата да е описал първо основните му клинични характеристики.
Крузон е живял между края на 1800 -те и началото на 1900 -те години, точно от 1874 до 1938 г. Първоначално, за да определи синдрома, който по -късно е получил името му, той използва термина черепно -лицева дизостоза.
Причини
Синдромът на Crouzon възниква в резултат на мутация в гена FGFR2, разположен на хромозома 10, или гена FGFR3, разположен на хромозома 4.
FGFR е английската акроним за Рецептор на фактор на растежа на фибробластите, което в превод на италиански е: Рецептор за фактора на растежа на фибробластите.
Функционалната роля на гените FGFR2 и FGFR3 е да произвеждат рецепторен протеин, който от своя страна има задачата да регулира узряването и ембрионалното развитие на костната тъкан.
Според теориите на изследователите, мутациите в FGFR2 и FGFR3 биха хиперстимулирали същите тези гени, които, след като бъдат по-активни, биха предизвикали ранно съзряване на някои костни тъкани, включително тези, съставляващи черепа.
ГЕНЕТИКА
Генетичните мутации, отговорни за синдрома на Крузон, могат да бъдат наследствени или да възникнат спонтанно след зачеването.
В първия случай болестното състояние - което лекарите също наричат наследствен синдром на Крузон - има всички характеристики на автозомно доминантно генетично заболяване (или наследствено доминиращо заболяване). За начинаещ читател на генетиката това означава, че:
- Заболяването и неговите симптоми се проявяват и при наличието само на един мутирал генен алел (няма значение дали идва от майката или от бащата), тъй като последният е доминиращ над здравия.
- Родител, носител на мутацията, е достатъчен, за да има болестта в част от потомството.
- Вероятността да се роди болно дете от двойка, в която само един от двата компонента носи мутацията, е 50%.
Във втория случай обаче болестното състояние - което експертите посочват с терминологията на ненаследствения синдром на Крузон - е резултат от аномално спорадично събитие, което променя ДНК по време на ембрионалния растеж на плода.
Обобщение на значението на термините наследствен, автозомна и доминантна
- Наследствено: означава, че родителите предават генетичната промяна, отговорна за болестта, на потомството (т.е. на децата).
- Автозомно: това означава, че мутацията, отговорна за болестта, се намира в несексуална хромозома, следователно автозомна.
- Доминиращ: означава, че болестта причинява симптоми и признаци, дори когато само един алел на отговорния ген е мутирал. По -просто казано, сякаш алелът с мутацията има по -голяма сила от здравия алел.
ЕПИДЕМИОЛОГИЯ
Според някои оценки на честотата на синдрома на Крузон, около едно на 60 000 деца ще се роди с това рядко състояние.
Синдромът на Крузон представлява 4,5% от случаите на краниосиностоза.
Симптоми и усложнения
Пациентите със синдром на Crouzon имат много специфична картина на симптомите, която обикновено се състои от:
- Проблеми, свързани с краниосиностоза, включително:
-

От https://en.wikipedia.org/wiki/Plagiocephaly Брахицефалия, което е притискане на задната част на главата. Следва преждевременно сливане на короналните черепни конци (коронална краниосиностоза).
Ако не се лекува, това може да повлияе на растежа на мозъка и развитието на когнитивните способности.
Те представляват „алтернатива на брахицефалията: тригоноцефалия (сливане на метопичния шев), долихоцефалия (сливане на сагиталния шев) и плагиоцефалия (сливане на коронарните конци). - Екзофталм, което е терминът за изпъкналостта на очните ябълки. Това може да означава наличие на проблеми със зрението.
- Очен хипертелоризъм, тоест очи, които са прекалено далечни един от друг. С екзофталмоса може да влоши зрителните проблеми.
- Деформиран нос, обикновено под формата на клюн. Ако е тежка или не се лекува хирургично, тази аномалия може да доведе до проблеми с дишането или до същите симптоми като синдрома на обструктивна сънна апнея.
- Повишено вътречерепно налягане. Известен е още като интракраниална хипертония. Наличието му се обяснява с факта, че мозъчните структури нямат подходящото пространство за растеж.
Обикновено открита в средата на късното детство, интракраниалната хипертония е потенциална причина за главоболие, повръщане и болка в очите. - Хидроцефалия, което е резултат от увеличаване на цереброспиналната течност, съдържаща се в субарахноидалното пространство и в мозъчните вентрикули.
- Малформация на Арнолд-Киари (или синдром на Арнолд-Киари). Това е деформация, разположена в основата на черепа.
* Хидроцефалията и малформацията на Арнолд-Киари обикновено са две усложнения, които възникват при липса на адекватно лечение. - Аномалии в долната и горната челюст.
Първият има по -малки размери от нормалното, докато вторият има тенденция да стърчи навън. Всичко това променя формата на небцето и зъбното скеле (липса на някои зъби и т.н.), с последици (понякога дори сериозни) върху фонацията и върху дъвчене.
Някои пациенти се раждат с цепка на устната (цепка на устната) или с цепнатина на небцето.
- Проблеми със слуха.
55% от пациентите със синдром на Крузон са родени без ушни канали или с големи аномалии в тях. Това води до липсващ или силно намален акустичен капацитет.
Някои субекти развиват набор от проблеми със слуха в зряла възраст, дължащи се на типичната клинична картина на синдрома на Мениер.
- Ставни проблеми в областта на шията.
Те засягат 30% от случаите на синдром на Crouzon.
- Кожни аномалии.
Налице са пациенти с мутирал FGFR3-поддържан синдром на Crouzon acanthosis nigricans, дерматоза, характеризираща се с увеличаване на дебелината (хиперкератоза) и потъмняване (хиперпигментация) на кожата.

Две други анатомични аномалии, свързани (макар и рядко) със синдрома на Crouzon
- Патентен артериален канал
- Коарктация на аортата
КРУЗОНОВ СИНДРОМ И IQ
Благодарение и на сегашните възможности за лечение на краниосиностоза, днес 97% от пациентите със синдром на Крузон имат „нормална интелигентност“.
Диагностика
Опитен педиатър може да бъде в състояние да диагностицира синдрома на Crouzon само чрез физически преглед.
При наличие на съмнение или недоумение, следното е от основно значение за достигане до точно заключение:
- Радиологични изображения, предоставени от рентгенови лъчи или CT сканиране на главата
- Генетичен тест, целящ да търси някакви ДНК мутации.
ОБЕКТИВЕН ИЗПИТ
Физическият преглед се състои в точен анализ на главата и аномалиите, присъстващи върху нея.
Черепните деформации, предизвикани от краниосиностоза (например брахицефалия), са сред най -характерните клинични признаци на синдрома на Крузон и върху които лекарят основава част от диагностичните си заключения.
РАДИОЛОГИЧНИ ИЗСЛЕДВАНИЯ
Рентгеновите лъчи и CT сканирането на главата показват кои черепни конци са се слели преждевременно.
Краниосиностозата, която характеризира синдрома на Crouzon, засяга коронарните конци, поради което констатираното сливане на нивото на последния е много често решаваща информация за диагностични цели.
ГЕНЕТИЧЕСКИ ИЗСЛЕДВАНЕ
Освен че показва дали ДНК има мутации, генетичното изследване помага да се идентифицира точния ген, който причинява синдрома на Crouzon, независимо дали FGFR2 или FGFR3.
Лечение
Днес носителите на синдрома на Crouzon могат да разчитат на различни лечения, в зависимост от тежестта на състоянието и симптомите.
Всъщност лекарите са осигурили:
- Хирургия за разрешаване на краниосиностоза и нейните симптоми.
- Акустични помощни средства, в случай на проблеми със слуха.
- Терапии за подобряване на езиковите умения.
- Хирургични терапии за подобряване на аномалиите в горната и долната челюст.
- Операция, известна като трахеостомия, за решаване на проблеми с дишането.
Моля обърнете внимание: Синдромът на Крузон е болестно състояние, което произтича от "генетична промяна на ДНК, невъзможна за лечение. Така че всъщност лекарите лекуват болестта само от симптоматична гледна точка.
ХИРУРГИЯ ЗА КРАНИОСИНОСТОЗА
Терапевтичните цели на хирургичната операция са две:
- Осигурете на мозъчните структури и очите пространството, от което се нуждаят, за да се развият и функционират най -добре.
- Придайте на главата нормална форма, след което решете проблема с брахицефалията.
Хирурзите имат възможност да извършат операцията по два различни начина (или подходи): чрез „традиционна хирургична операция - наричана още„ отворена ” - или чрез„ ендоскопска хирургична операция.
"Отворената операция" включва "извършване на" разрез на главата, чрез който опериращият лекар извлича костта или неправилно оформените черепни кости, които трябва да бъдат ремоделирани. В края на ремоделирането хирургът поставя отново костните структури, предварително екстрахирани, и затваря разреза с конци.
Ендоскопската хирургия, от друга страна, включва използването на ендоскоп и практикуването на много малък разрез на главата, чрез който опериращият лекар вкарва самия ендоскоп.
Ендоскопът всъщност е тънка и гъвкава тръба, оборудвана с оптична камера (в края поставена в черепа) и свързана с монитор. Чрез този конкретен инструмент и изображенията, които той прожектира върху монитора, хирургът е в състояние да отдели преждевременно сливащите се черепни конци, със забележителна прецизност и без да прибягва до кожни разрези и костни екстракции.
Според експерти най -доброто време за извършване на операцията е през много ранна детска възраст (първите 12 месеца от живота), тъй като костите се оформят по -лесно.
Трябва обаче да се помни, че колкото по -млад е пациентът, толкова по -голям е рискът от повторение на същите черепни конци (рецидив) .В случай на рецидив операцията трябва да се повтори.
Според някои статистически изследвания 10-20% от много младите пациенти, подложени на операция с краниосиностоза, трябва да се подложат на втора операция поради рецидив.
ЛЕЧЕНИЕ НА АКУСТИЧНИ ПРОБЛЕМИ
Освен че предписват използването на слухови апарати, лекарите препоръчват и периодични слухови проверки, тъй като това е най -добрият начин за предотвратяване на влошаване на съществуващите проблеми.
ХИРУРГИЧНА ТЕРАПИЯ ЗА ЧЕЛУПА И ЧЕРЕВИ АНОМАЛИИ
Лечението на максиларни и мандибуларни аномалии включва операция за пренареждане на максилата и / или долната челюст, някои стоматологични лечения за подреждането на зъбните дъги и операцията за разрешаване на цепнатината на устната и / или цепнатината на небцето.
ТРАХЕОСТОМИЯ
Трахеостомията е хирургичната операция, чрез която лекарят създава на нивото на шията (където трахеята преминава) проход за въздуха, предназначен за белите дробове. Това позволява на тези, които са подложени на тази операция, да дишат отново и правилно.
За да пренесете въздуха в белите дробове, се нуждаете от малка тръба, наречена трансхеостомична тръба, която е с правилния размер за вмъкване в трахеята.
Прогноза
Като цяло прогнозата зависи от тежестта на краниосиностозата: ако последната се лекува с добри резултати, пациентите със синдром на Crouzon могат да се радват на почти нормален живот.









.jpg)