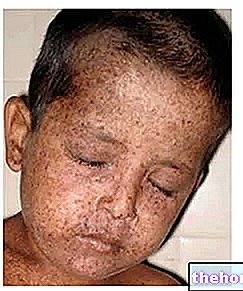
Това заболяване дължи името си на американския ендокринолог, който го е открил: Фредерик Кросби Бартър.Годишната честота е оценена на 1 /830 000.
Има няколко варианта на синдрома на Бартър, чието предаване, макар и все още автозомно, може да варира от рецесивно до доминиращо в зависимост от случая.
Ако не бъде своевременно диагностициран и лекуван, синдромът на Бартър може сериозно да компрометира развитието, растежа и качеството на живот на пациента. Освен това в особено тежки случаи продължителността на живота се намалява значително.
Моля обърнете внимание
Синдромът на Бартър НЕ трябва да се бърка със синдрома на Шварц-Бартър, заболяване, характеризиращо се с „нарушена секреция на антидуиретичен хормон (ADH), известен също като синдром на неподходяща секреция на ADH (SIADH).
.което се случва на нивото на бримката на Henle се дължи на "промяна в синтеза на някои рецептори на канал / транспортер (конкретни протеини, които транспортират йони от различно естество), разположени в тази" област на бъбрека. Това явление се причинява чрез поредица генетични мутации, засягащи гените, кодиращи гореспоменатите конкретни протеини.
Различните варианти на синдрома на Бартър се разграничават според засегнатия ген. По -подробна информация за това може да бъде намерена в следващата глава.
.
Следователно следващата таблица ще покаже различните варианти на синдрома, включените мутирали гени, протеините (канални рецептори / транспортери), за които те кодират, и клиничното представяне на въпросния вариант.
Вариант
Мутирал ген
Включен канал / транспортер
Клинична презентация
Синдром на Бартър тип I
Ген SLC12A1
NKCC2 (котранспортер натрий-калий-хлор или Na + / K + / 2Cl-)
Пренатален (или инфантилен) синдром на Бартър
Синдром на Бартър тип II
Ген KCNJ1
ROMK (калиев канал на външната медула на бъбрека)
Пренатален (или инфантилен) синдром на Бартър
Синдром на Бартър тип III
Ген CLNKb
CLCNKb (хлорен канал тип Kb)
Класически синдром на Бартър
Синдром на Бартър тип IV или IV А
Ген BSND
Бартина (бета субединица на хлорни канали тип Ka и Kb)
Пренатален (или инфантилен) синдром на Бартър и сензоневрална загуба на слуха
Синдром на Бартър тип IV В
CLCNKa и CLCNKb гени
CLCNKa (хлорен канал тип Ka) и CLCNKb
Пренатален (или инфантилен) синдром на Бартър и сензоневрална глухота
Бартъров синдром тип V
CASR ген
CaSR (чувствителен към калций рецептор)
Синдром на Бартър с хипокалциемия
Както се вижда от таблицата, въпреки наличието на пет генетични варианта, не е възможно да се разграничат толкова клинични форми; всъщност се разграничават само четири: пренатален или инфантилен синдром на Бартър (тип I и II), класически синдром на Бартър (тип III), пренатален или инфантилен синдром на Бартър, свързан със сензоневрална глухота (тип IV А и IV В; някои източници обаче те групират тези варианти заедно с тип I и II) и накрая, синдром на Бартър с хипокалциемия (тип V).
Знаете ли, че ...
Като се има предвид наличието на вариант IV (или IV A) и вариант IV B на синдрома на Бартър, някои източници разглеждат като цяло шест варианта на синдрома на Бартър. Други източници обаче разглеждат вариант IV В като подтип на вариант IV и , поради тази причина, обмисляйте съществуването само на пет генетични варианта на синдрома на Бартър.
Варианти от тип I, II, III, IV и IV B са автозомно рецесивно предавани заболявания, което означава, че за да се прояви синдромът, индивидът трябва да притежава и двата мутирали алела, наследявайки ги от родителите, които следователно ще бъдат здрави носители. на синдрома, от друга страна, е автозомно доминантно предаване, това означава, че за да се проявят симптомите, е достатъчно пациентът да притежава един мутирал алел, който следователно може да бъде наследен дори само от един (също болен ) на двамата родители.
Псевдосиндром на Бартър
Псевдосиндромът на Бартър е състояние, характеризиращо се със симптоми, подобни на тези, предизвикани от синдрома на Бартър, но чиято причина трябва да се открие в злоупотребата с диуретични лекарства като фуроземид.
Синдром на Gitelman
Този синдром се причинява от локализирана мутация върху гена SLC12A3, който кодира натриево-хлорния котранспортер (NCC). Поради тази мутация - предадена по автозомно -рецесивен начин - пациентът претърпява нарушение на реабсорбцията на натрий, хлор и калий на нивото на дисталната извита тубула, за разлика от синдрома на Бартър, при който увреждането на резорбцията се локализира в „Въпреки това , Синдромът на Gitelman може да породи симптоми, подобни на тези на синдрома на Bartter, поради което в клиничната практика понякога може да бъде трудно да се разграничат двете заболявания.
, хипохлоремия и метаболитна алкалоза, които могат да бъдат свързани с хиперренинемия (повишен ренин в кръвта) и хипералдостеронизъм. Ясно е, че всички тези състояния от своя страна могат да доведат до поредица от симптоми, способни да нарушат качеството на живот на пациента (например гадене, повръщане, замаяност, слабост, главоболие, хипотония и др.).
В допълнение към казаното досега, всеки вариант може да породи специфични прояви и симптоми, тясно свързани с мутиралия ген и с последващото включване на канала или котранспортера, за който този ген кодира. Следователно, типичните симптоми и прояви, свързани с всяка от петте различни форми на синдрома на Бартър, ще бъдат описани накратко по -долу.
Синдром на Бартър тип I
При синдром на Бартър тип I мутациите засягат гена, кодиращ котранспортера натрий-калий-хлор, присъстващ на бримката на Henle.Поради компрометираната реабсорбция, възниква хиповолемия поради загуба на соли. В същото време, тъй като реабсорбцията на калций също е свързана с активността на гореспоменатия котранспортер, ние сме свидетели на появата на хиперкалциурия, Всичко това може да доведе до появата на нефрокалциноза. Възможно е също да изпитате хипермагнезурия. Полихидрамнион, вторичен на феталната полиурия, може да се развие в пренаталния период.
Синдром на Бартър тип II
Синдромът на Бартър тип II се причинява от мутация в гена, който кодира калиевия канал на надбъбречната медула. Проявите и симптомите са сходни с тези на вариант I и също в този случай може да се срещне полихидрамнион, вторичен на феталната полиурия. Въпреки това, на ранен етап, новороденото може да получи преходна хиперкалиемична метаболитна ацидоза. Това състояние след това се развива към характерната клинична картина на синдрома на Бартър.
Синдром на Бартър тип III
Известен също като класически синдром на Бартър, вариант III на заболяването се причинява от мутации в гена, кодиращ хлорния канал от типа Kb.Тъй като хлорните канали от типа Ка се запазват в тази форма, симптомите са склонни да бъдат по -леки, въпреки че все още присъстват. По принцип няма нефрокалциноза.
Синдром на Бартър тип IV и IV B
И при двата типа вариант IV има участие на гени, участващи в правилния синтез на Ka и Kb хлорните канали. Тъй като и двата канала са компрометирани, симптомите са по -тежки, отколкото при вариант III на синдрома.. Бебетата първоначално могат да покажат клинична картина, която имитира хипоалдостеронизъм, но която след това се развива към хипокалиемична метаболитна алкалоза, когато тялото се опитва да компенсира липсата на активност на гореспоменатите калциеви канали. Характерно за варианти IV и IV B на синдрома на Бартър е появата на сензоневрална глухота.
Бартъров синдром тип V
Вариант V на синдрома на Бартър се причинява от мутация, засягаща гена, кодиращ чувствителния на калций рецептор, участващ в инхибирането на реабсорбцията на вода и различни йони, като калций, калий и натрий. последваща хиперкалциурия, свързана с характерните симптоми на синдрома на Бартър.
Знаете ли, че ...
Варианти I, II, IV и IV B на синдрома на Бартър - както и с името на пренаталния синдром на Бартър - понякога се наричат също синдром на хипепростагландин Е2, тъй като се характеризират с повишаване на плазмените нива на този простагландин.
- насочени към установяване наличието и концентрацията на електролити (натрий, калий, хлорид, магнезий, бикарбонат, калций) и специфични вещества (ренин и алдостерон) в плазмата и / или урината.Окончателната диагноза е възможна само при извършване на специфични генетични тестове.
Диференциалната диагноза, от друга страна, трябва да бъде поставена срещу псевдосиндрома на Бартър, синдрома на Гителман, кистозна фиброза и цьолиакия.
В случаите, когато съществува определен риск (например родители със здрави и / или болни носители), че новороденото може да прояви болестта, е възможна и пренатална диагностика.
от:
- Добавки от минерални соли (по -специално, но не изключително, калий) с цел компенсиране на липсата на реабсорбция;
- Нестероидни противовъзпалителни средства (НСПВС), като например индометацин.Тези лекарства се прилагат с цел намаляване на прекомерно високи нива на простагландин Е2;
- Калий-съхраняващи диуретици (прилагани за намаляване на отделянето на калий в урината).
В най -тежките случаи и / или при стресови състояния (поява на други заболявания, хирургични интервенции и т.н.), попълването на калий и други минерални соли може да се извърши интравенозно, разбира се, подобна операция трябва да се извърши и от здравето персонал, специализиран.

-cos-cause-e-terapia.jpg)