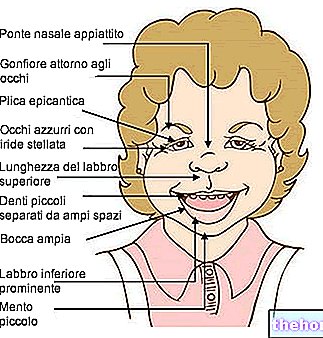
Какво е фенилкетонурия
Там фенилкетонурия (P.K.U.) това е автозомно рецесивно наследствено метаболитно заболяване, което засяга 1 на 10 000 индивида и изглежда се среща по -често при хомозиготност, отколкото при хетерозиготи.
Принадлежаща към групата на хиперфенилаланинемия, фенилкетонурията значително компрометира метаболизма на фенилаланин и по -специално неговия преобразуване в тирозин; фенилкетонурията се разпознава по повишените нива на фенилаланин и някои производни в урината (фенилпируват, фенилацетат, фенилактат и фенилацетилглутамин).
Най -сериозното усложнение на фенилкетонурията е умствено забавяне.
Фенилаланин, тирозин и производни
Фенилаланинът е незаменима аминокиселина и съставлява по -голямата част от хранителните протеини; може да се преобразува от ензима фенилаланин хидроксилаза в тирозин (чрез добавяне на хидроксилна група -ОН). От своя страна тирозинът е аминокиселина -предшественик за синтеза на:
- L-DOPA (междинно съединение за синтез на допамин)
- Епинефрин
- Норадреналин (всички невротрансмитери).

Механизъм на фенилкетонурия (P.K.U.)
Както се очакваше, при фенилкетонурия, поради една или повече (общо 6) хромозомни мутации, експресията (оттам и метаболитната активност) на фенилаланин хидроксилаза е практически нулева. Тези промени могат да бъдат от различен вид (от „неправилни“ промени до „сплайсинг“ дефекти или дори „частични заличавания“), но важното е, че поради тази ензимна неефективност нивата на фенилаланин в кръвта (които обикновено са 1 mg / 100 ml) при ДОМИНАТНА фенилкетонурия те лесно достигат количества дори 50 пъти по -високи.
Функциониране на ензима фенилаланин хидроксилаза: За производството на тирозин (+ дихидробиоптерин), фенилаланин хидроксилазата изисква: фенилаланин, кислород и тетрахидробиоптерин (редуциран птеридин, който действа като кофактор); реакцията също е обратима и дихидробиоптеринът може да се преобразува отново (благодарение на ензима дихидроптерин редуктаза) в тетрахидробиоптерин.
Усложнения
Фенилкетонурията може да доведе до повече или по -малко тежки усложнения въз основа на тежестта на патологичната проява и навременността на диагнозата; като наследствена патология, фенилкетонурията се отличава с:
- Доминиращ, следователно характеризиращ се с ПЪЛНО бездействие на ензима фенилаланин хидроксилаза
- Рецесивен, при който само 30% от общото ензимно наследство е активно.
Усложненията на фенилкетонурията се дължат и са пряко пропорционални на метаболитното натрупване на фенилаланин, неговите производни и намаления синтез на тирозин.При патология излишъкът от фенилаланин се филтрира сравнително ефективно от бъбрека, който го абсорбира само частично, отстранявайки го с урината ; обаче, постоянството на нивата на хиперфенилаланинемия определя метаболитна реакция на молекулярно ПРЕВЪРНЕНИЕ в фенилпировиновата киселина и / или други производни, които са по -лесни за източване (фенилпируват, фенилацетат, фенилактат).
Това, което усложнява фенилкетонурията, е токсичността на фенилаланин, фенилпировиновата киселина и нейните производни към централната нервна система (ЦНС). Прекомерното им присъствие в развитието на мозъка неумолимо определя форма на умствена изостаналост.
NB. Плазмените концентрации на другите аминокиселини са леко намалени, вероятно поради обратна връзка за чревна абсорбция или бъбречна тубулна реабсорбция.
Увреждането на мозъка, като сериозно усложнение на фенилкетонурията, се причинява от изваждането на други незаменими аминокиселини в протеосинтезата, по -специално при образуването на полирибозоми, миелин, норадреналин и серотонин. Фенилкетонурия - не се вижда веднага след раждането, но след няколко години - ако не се лекува, изисква хоспитализация на детето и е напълно необратима.
Разширената фенилкетонурия също може да бъде ясно видима с просто око; високите концентрации на фенилаланин, инхибиращ ензима тирозиназа, значително влошават синтеза на меланин, като намаляват пигментацията на кожата и косата; освен това, натрупването на фенилацетат в косата и кожата придава на фенилкетонуриците силен и неприятен "мирис на мишка".